A function for comparing and ranking predicted means with Tukey's Honest Significant Difference (HSD) Test.
Usage
multiple_comparisons(
model.obj,
classify,
sig = 0.05,
int.type = "ci",
trans = NULL,
offset = NULL,
power = NULL,
decimals = 2,
descending = FALSE,
groups = TRUE,
adjust = "tukey",
by = NULL,
plot = FALSE,
label_height = 0.1,
rotation = 0,
save = FALSE,
savename = "predicted_values",
...
)Arguments
- model.obj
An
asreml,aov,lm,lme(nlme::lme()) orlmerMod(lme4::lmer()) model object.- classify
Name of predictor variable as string.
- sig
The significance level, numeric between 0 and 1. Default is 0.05.
- int.type
The type of confidence interval to calculate. One of
ci,tukey,1se,2se, ornone. Default isci.- trans
Transformation that was applied to the response variable. One of
log,sqrt,logit,power,inverse, orarcsin. Default isNULL.- offset
Numeric offset applied to response variable prior to transformation. Default is
NULL. Use 0 if no offset was applied to the transformed data. See Details for more information.- power
Numeric power applied to response variable with power transformation. Default is
NULL. See Details for more information.- decimals
Deprecated. Rounding is now controlled via the
decimalsargument ofprint.mct().- descending
Logical (default
FALSE). Order of the output sorted by the predicted value. IfTRUE, largest will be first, through to smallest last.- groups
Logical (default
TRUE). IfTRUE, the significance letter groupings will be calculated and displayed. This can get overwhelming for large numbers of comparisons, so can be turned off by setting toFALSE.- adjust
The method used to adjust p-values for multiple comparisons. Either
"tukey"(default, Tukey's HSD) or any method accepted bystats::p.adjust()("bonferroni","holm","hochberg","hommel","BH"(or"fdr"),"BY", or"none"). See Details.- by
A character vector of column name(s) in the predictions over which to split comparisons. Comparisons are run independently within each level (or combination of levels) of the
byvariable(s); no p-values are pooled or adjusted across groups. DefaultNULL. See Details.- plot
Automatically produce a plot of the output of the multiple comparison test? Default is
FALSE. This is maintained for backwards compatibility, but the preferred method now is to useautoplot(<multiple_comparisons output>). Seeautoplot.mct()for more details.- label_height
Height of the text labels above the upper error bar on the plot. Default is 0.1 (10%) of the difference between upper and lower error bars above the top error bar.
- rotation
Rotate the text output as Treatments within the plot. Allows for easier reading of long treatment labels. Number between 0 and 360 (inclusive) - default 0
- save
Logical (default
FALSE). Save the predicted values to a csv file?- savename
A file name for the predicted values to be saved to. Default is
predicted_values.- ...
Other arguments passed internally to model-specific prediction methods.
Value
An object of class mct (a list with class attributes) containing:
- predictions
A data frame with predicted means, standard errors, confidence interval upper and lower bounds, and significant group allocations
- pairwise_pvalues
A symmetric matrix of adjusted p-values for all pairwise comparisons (Tukey's HSD by default, otherwise adjusted by the
adjustmethod). Whenbyis supplied, a named list of such matrices, one per subgroup- hsd
The Honest Significant Difference value(s) used in the comparisons when
adjust = "tukey". Either a single numeric value (if constant across comparisons) or a matrix (if it varies by comparison).NULLwhenadjustis not"tukey"- sig_level
The significance level used (default 0.05)
- comparison_method
The p-value adjustment method used (the value of
adjust)- aliased
Character vector of aliased treatment levels (only present if some predictions are aliased)
Details
Offset
Some transformations require that data has a small offset to be
applied, otherwise it will cause errors (for example taking a log of 0, or
the square root of negative values). In order to correctly reverse this
offset, if the trans argument is supplied, a value should also be supplied
in the offset argument. By default the function assumes no offset was
required for a transformation, implying a value of 0 for the offset
argument. If an offset value is provided, use the same value as provided in
the model, not the inverse. For example, if adding 0.1 to values for a log
transformation, add 0.1 in the offset argument.
Power
The power argument allows the specification of arbitrary powers to be back transformed, if they have been used to attempt to improve normality of residuals.
P-value adjustment (adjust)
By default (adjust = "tukey") the function uses Tukey's HSD, which is exact
for the complete set of all pairwise comparisons. Alternatively, adjust may
be any method accepted by stats::p.adjust(). In that case a matrix of raw
two-sided t-test p-values is computed first and the chosen adjustment is
applied to the lower-triangle vector of those raw p-values, avoiding the
double-adjustment that would occur if Tukey-adjusted p-values were passed to
p.adjust(). Bonferroni and Holm control the family-wise error rate and are
valid under arbitrary dependence; BH/BY (FDR) are valid under positive
dependence, which pairwise comparisons satisfy. The returned
$pairwise_pvalues always holds the adjusted p-values for the chosen method
(the Tukey p-values when adjust = "tukey"). When adjust is not "tukey",
$hsd is NULL, as an HSD value is only meaningful for Tukey's test.
Grouped comparisons (by)
When by is supplied, the predictions are split into subgroups defined by
the level(s) of the by variable(s) and the comparison procedure is run
independently within each subgroup. Each subgroup is treated as a separate
family of comparisons: there is no pooling and no cross-group p-value
adjustment, and letter groupings restart within each subgroup. When by is
used, $pairwise_pvalues (and $hsd where applicable) are returned as named
lists with one element per subgroup, and autoplot() facets the plot by the
by variable(s) by default. by must leave at least one classify factor
to compare within each subgroup, so a single-factor classify cannot be
split with by.
Confidence Intervals & Comparison Intervals
The function provides several options for confidence intervals via the int.type argument:
ci(default): Traditional confidence intervals for individual means. These estimate the precision of each individual mean but may not align with the multiple comparison results. Non-overlapping traditional confidence intervals do not necessarily indicate significant differences in multiple comparison tests.tukey: Tukey comparison intervals that are consistent with the multiple comparison test. These intervals are wider than regular confidence intervals and are designed so that non-overlapping intervals correspond to statistically significant differences in the Tukey HSD test. This ensures visual consistency between the intervals and letter groupings.1seand2se: Intervals of ±1 or ±2 standard errors around each mean.none: No confidence intervals will be calculated or displayed in plots.
By default, the function displays regular confidence intervals (int.type = "ci"),
which estimate the precision of individual treatment means. However, when
performing multiple comparisons, these regular confidence intervals may not
align with the letter groupings from Tukey's HSD test. Specifically, you may
observe non-overlapping confidence intervals for treatments that share the
same letter group (indicating no significant difference).
This occurs because regular confidence intervals and Tukey's HSD test serve different purposes:
Regular confidence intervals estimate individual mean precision
Tukey's HSD controls the family-wise error rate across all pairwise comparisons
To resolve this visual inconsistency, you can use Tukey comparison intervals
(int.type = "tukey"). These intervals are specifically designed for multiple
comparisons and will be consistent with the letter groupings: non-overlapping
Tukey intervals indicate significant differences, while overlapping intervals
suggest no significant difference.
The function will issue a message if it detects potential inconsistencies between non-overlapping confidence intervals and letter groupings, suggesting the use of Tukey intervals for clearer interpretation. For multiple comparison contexts, Tukey comparison intervals are recommended as they provide visual consistency with the statistical test being performed and avoid the common confusion where traditional confidence intervals don't overlap but groups share the same significance letter.
Supported model types
The comparison functions (multiple_comparisons(), pairwise_comparisons()
and reference_comparisons()) work with any model for which a
get_predictions() method is defined. These are currently:
| Model class | Fitted by | Notes |
aov, lm | stats::aov(), stats::lm() | Fixed-effects linear models. |
aovlist | stats::aov() with an Error() term | Multi-stratum aov; gives comparison-specific (matrix) degrees of freedom. |
lme | nlme::lme() | Linear mixed model. |
lmerMod | lme4::lmer(), lme4breeding::lmebreed() | Linear mixed model. lmebreed() (relationship-based) models also carry class lmerMod; comparisons target the fixed-effect means with Kenward-Roger degrees of freedom, and correctly reflect the relationship structure (validated against ASReml-R). |
lmerModLmerTest | lmerTest::lmer() | As lmerMod, with Satterthwaite degrees of freedom. |
asreml | ASReml-R asreml() | Linear mixed model (commercial; not on CRAN). |
afex_aov | afex aov_car() / aov_ez() / aov_4() | Factorial / repeated-measures ANOVA; gives comparison-specific (matrix) degrees of freedom. |
glmmTMB | glmmTMB glmmTMB() | Generalized linear mixed model. Predictions are on the link scale with asymptotic (infinite) degrees of freedom; supply trans to back-transform. |
mmes | sommer mmes() | Linear mixed model, via sommer's native predict(). SED from the prediction covariance; asymptotic (infinite) degrees of freedom (sommer provides none). |
ARTool (art) models are supported by resplot() but not by the comparison
functions: the aligned rank transform makes mean-based comparisons inappropriate.
Use ARTool::art.con() for contrasts on ART models instead.
sommer mmer models (the legacy interface) are supported by resplot() but
not by the comparison functions: current sommer provides no predict() method
for mmer. Refit with sommer::mmes() to use the comparison functions.
To add a new engine, write a get_predictions.<class>() method returning a
list with elements predictions, sed, df, ylab and aliased_names
(plus emmeans_grid for engines backed by emmeans::emmeans()), and add a
row to the table above.
References
Jørgensen, E. & Pedersen, A. R. (1997). How to Obtain Those Nasty Standard Errors From Transformed Data - and Why They Should Not Be Used.
See also
pairwise_comparisons() for testing a chosen subset of pairwise
differences as a tidy table, or reference_comparisons() for testing
treatments against a chosen reference or control. For guidance on choosing
between the two and on multiplicity adjustments, see
vignette("choosing-multiple-comparisons", "biometryassist").
Examples
# Fit aov model
model <- aov(Petal.Length ~ Species, data = iris)
# Display the ANOVA table for the model
anova(model)
#> Analysis of Variance Table
#>
#> Response: Petal.Length
#> Df Sum Sq Mean Sq F value Pr(>F)
#> Species 2 437.10 218.551 1180.2 < 2.2e-16 ***
#> Residuals 147 27.22 0.185
#> ---
#> Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
# Determine ranking and groups according to Tukey's Test (with Tukey intervals)
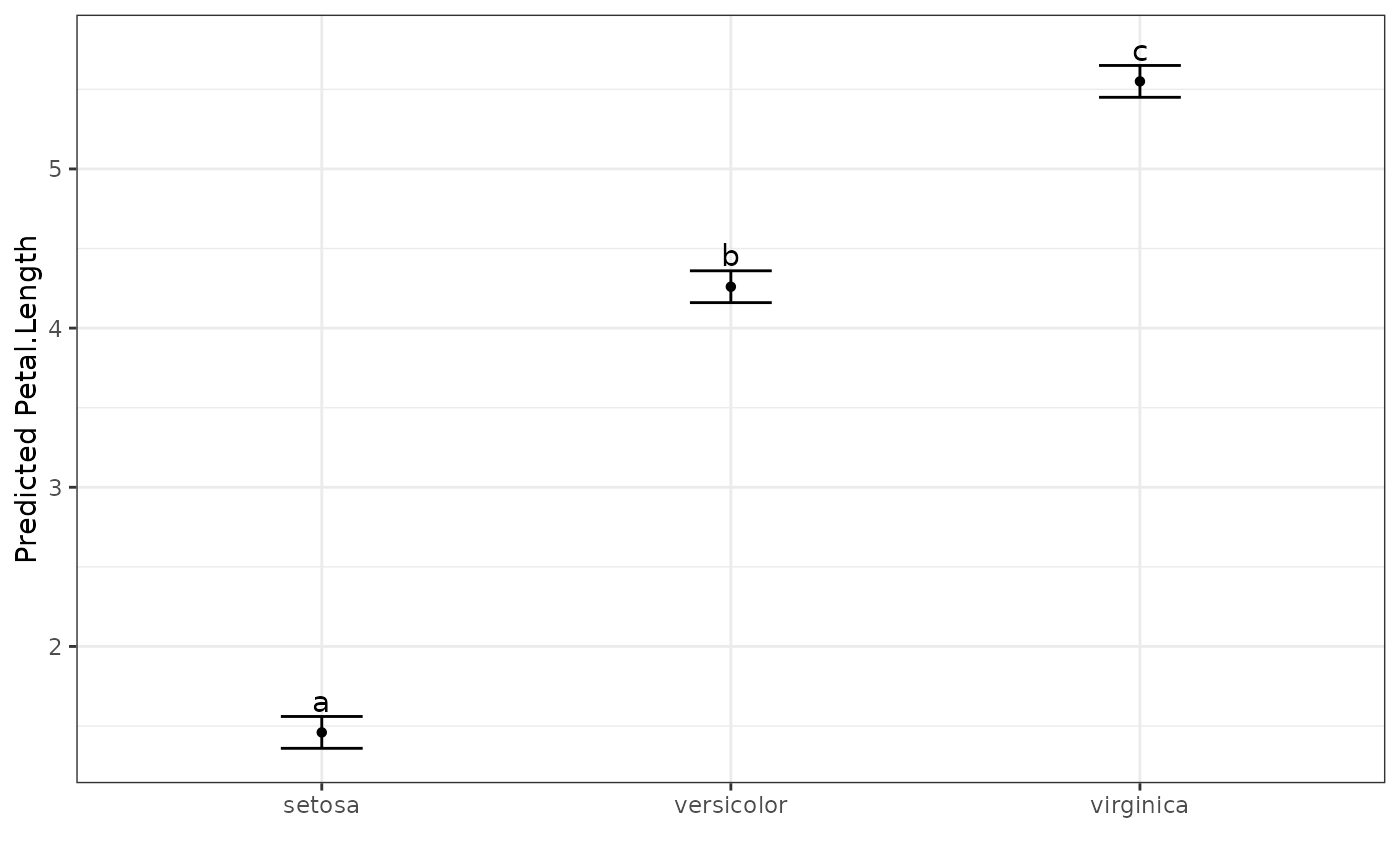
pred.out <- multiple_comparisons(model, classify = "Species")
# Display the predicted values table
pred.out
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: 0.20378
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci low up
#> 1 setosa 1.46 0.06 147 a 0.12 1.34 1.58
#> 2 versicolor 4.26 0.06 147 b 0.12 4.14 4.38
#> 3 virginica 5.55 0.06 147 c 0.12 5.43 5.67
# Access the p-value matrix
pred.out$pairwise_pvalues
#> setosa versicolor virginica
#> setosa 1.000000e+00 2.997602e-15 2.997602e-15
#> versicolor 2.997602e-15 1.000000e+00 2.997602e-15
#> virginica 2.997602e-15 2.997602e-15 1.000000e+00
# Access the HSD value
pred.out$hsd
#> [1] 0.20378
# Show the predicted values plot
autoplot(pred.out, label_height = 0.5)
 # Use traditional confidence intervals instead of Tukey comparison intervals
pred.out.ci <- multiple_comparisons(model, classify = "Species", int.type = "ci")
pred.out.ci
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: 0.20378
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci low up
#> 1 setosa 1.46 0.06 147 a 0.12 1.34 1.58
#> 2 versicolor 4.26 0.06 147 b 0.12 4.14 4.38
#> 3 virginica 5.55 0.06 147 c 0.12 5.43 5.67
# Plot without confidence intervals
pred.out.none <- multiple_comparisons(model, classify = "Species", int.type = "none")
autoplot(pred.out.none)
# Use traditional confidence intervals instead of Tukey comparison intervals
pred.out.ci <- multiple_comparisons(model, classify = "Species", int.type = "ci")
pred.out.ci
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: 0.20378
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci low up
#> 1 setosa 1.46 0.06 147 a 0.12 1.34 1.58
#> 2 versicolor 4.26 0.06 147 b 0.12 4.14 4.38
#> 3 virginica 5.55 0.06 147 c 0.12 5.43 5.67
# Plot without confidence intervals
pred.out.none <- multiple_comparisons(model, classify = "Species", int.type = "none")
autoplot(pred.out.none)
 # Use a different p-value adjustment instead of Tukey's HSD
multiple_comparisons(model, classify = "Species", adjust = "fdr")
#> Multiple Comparisons of Means: p-value adjustment = fdr
#> Significance level: 0.05
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci low up
#> 1 setosa 1.46 0.06 147 a 0.12 1.34 1.58
#> 2 versicolor 4.26 0.06 147 b 0.12 4.14 4.38
#> 3 virginica 5.55 0.06 147 c 0.12 5.43 5.67
# `by`: run the comparisons independently within each level of another factor.
# Here a 2 x 3 factorial - compare tension levels within each wool type.
m_wb <- aov(breaks ~ wool * tension, data = warpbreaks)
mc_by <- multiple_comparisons(m_wb, classify = "wool:tension", by = "wool")
mc_by
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: varies by comparison (see $hsd)
#>
#>
#> Predicted values:
#> wool tension predicted.value std.error df groups ci low up
#> 1 A M 24.00 3.65 48 a 7.33 16.67 31.33
#> 2 A H 24.56 3.65 48 a 7.33 17.22 31.89
#> 3 A L 44.56 3.65 48 b 7.33 37.22 51.89
#> 4 B H 18.78 3.65 48 a 7.33 11.45 26.11
#> 5 B L 28.22 3.65 48 a 7.33 20.89 35.55
#> 6 B M 28.78 3.65 48 a 7.33 21.45 36.11
autoplot(mc_by) # faceted by wool
# Use a different p-value adjustment instead of Tukey's HSD
multiple_comparisons(model, classify = "Species", adjust = "fdr")
#> Multiple Comparisons of Means: p-value adjustment = fdr
#> Significance level: 0.05
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci low up
#> 1 setosa 1.46 0.06 147 a 0.12 1.34 1.58
#> 2 versicolor 4.26 0.06 147 b 0.12 4.14 4.38
#> 3 virginica 5.55 0.06 147 c 0.12 5.43 5.67
# `by`: run the comparisons independently within each level of another factor.
# Here a 2 x 3 factorial - compare tension levels within each wool type.
m_wb <- aov(breaks ~ wool * tension, data = warpbreaks)
mc_by <- multiple_comparisons(m_wb, classify = "wool:tension", by = "wool")
mc_by
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: varies by comparison (see $hsd)
#>
#>
#> Predicted values:
#> wool tension predicted.value std.error df groups ci low up
#> 1 A M 24.00 3.65 48 a 7.33 16.67 31.33
#> 2 A H 24.56 3.65 48 a 7.33 17.22 31.89
#> 3 A L 44.56 3.65 48 b 7.33 37.22 51.89
#> 4 B H 18.78 3.65 48 a 7.33 11.45 26.11
#> 5 B L 28.22 3.65 48 a 7.33 20.89 35.55
#> 6 B M 28.78 3.65 48 a 7.33 21.45 36.11
autoplot(mc_by) # faceted by wool
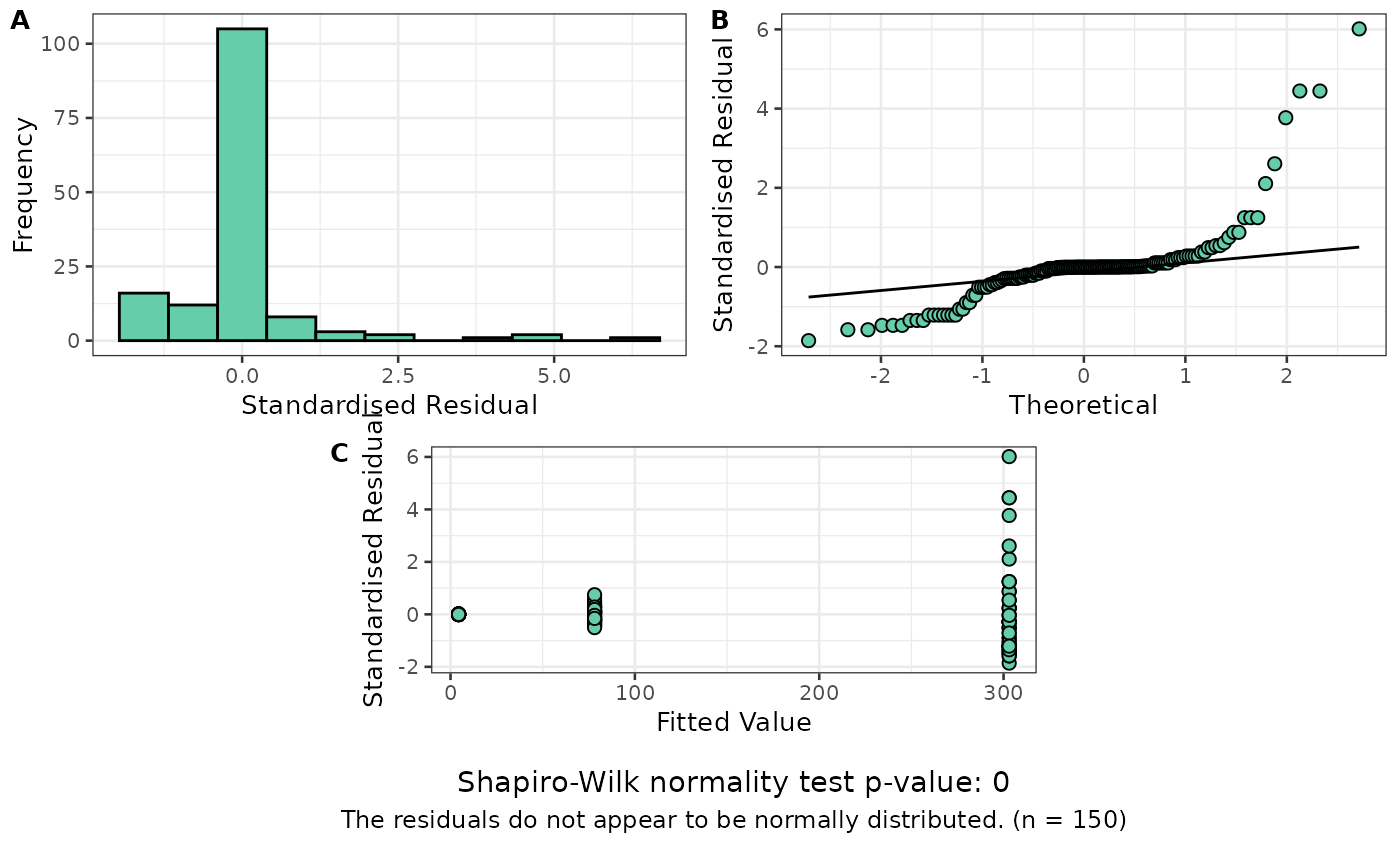
 # AOV model example with transformation
my_iris <- iris
my_iris$Petal.Length <- exp(my_iris$Petal.Length) # Create exponential response
exp_model <- aov(Petal.Length ~ Species, data = my_iris)
resplot(exp_model) # Residual plot shows problems
# AOV model example with transformation
my_iris <- iris
my_iris$Petal.Length <- exp(my_iris$Petal.Length) # Create exponential response
exp_model <- aov(Petal.Length ~ Species, data = my_iris)
resplot(exp_model) # Residual plot shows problems
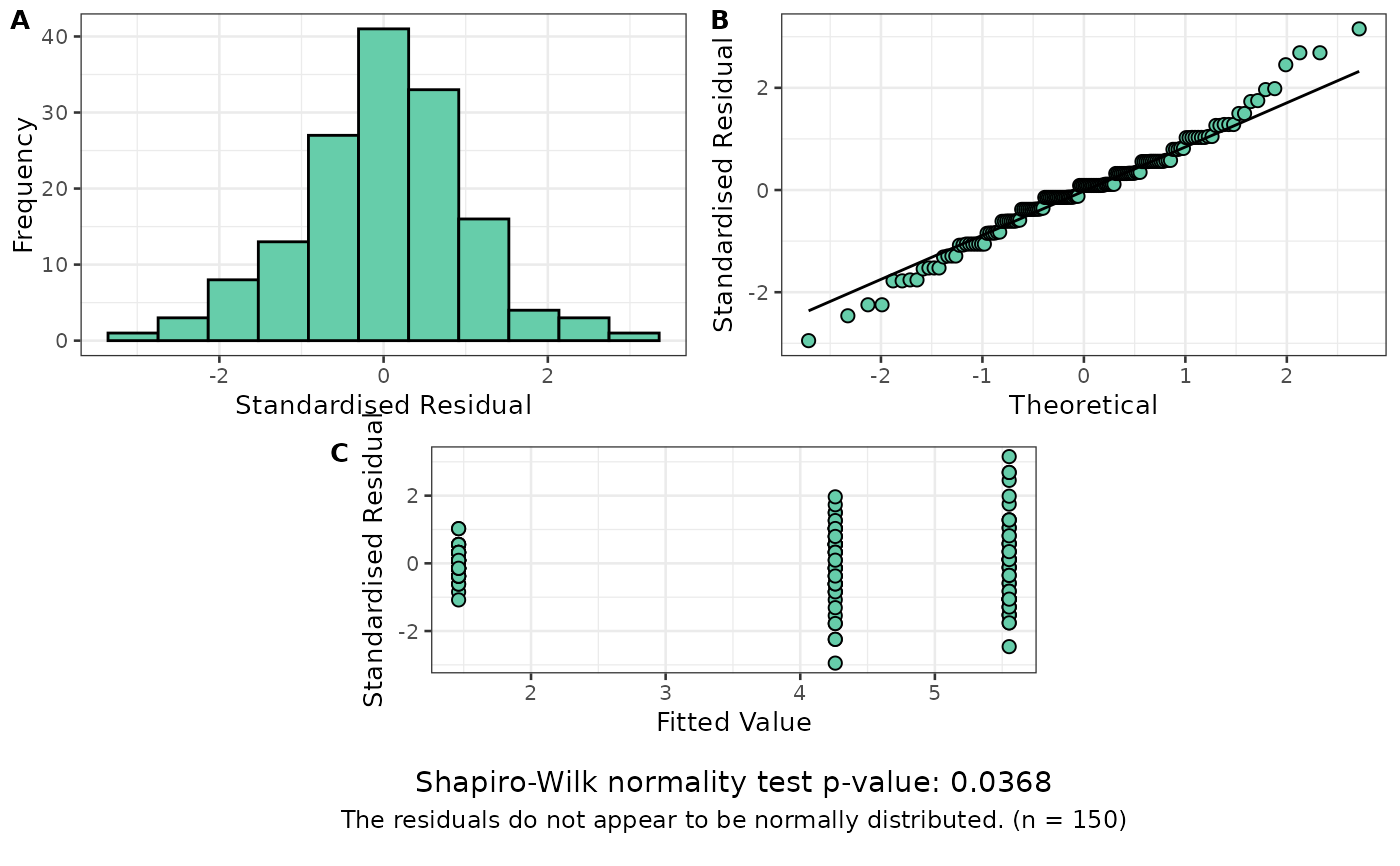
 # Fit a new model using a log transformation of the response
log_model <- aov(log(Petal.Length) ~ Species, data = my_iris)
resplot(log_model) # Looks much better
# Fit a new model using a log transformation of the response
log_model <- aov(log(Petal.Length) ~ Species, data = my_iris)
resplot(log_model) # Looks much better
 # Display the ANOVA table for the model
anova(log_model)
#> Analysis of Variance Table
#>
#> Response: log(Petal.Length)
#> Df Sum Sq Mean Sq F value Pr(>F)
#> Species 2 437.10 218.551 1180.2 < 2.2e-16 ***
#> Residuals 147 27.22 0.185
#> ---
#> Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
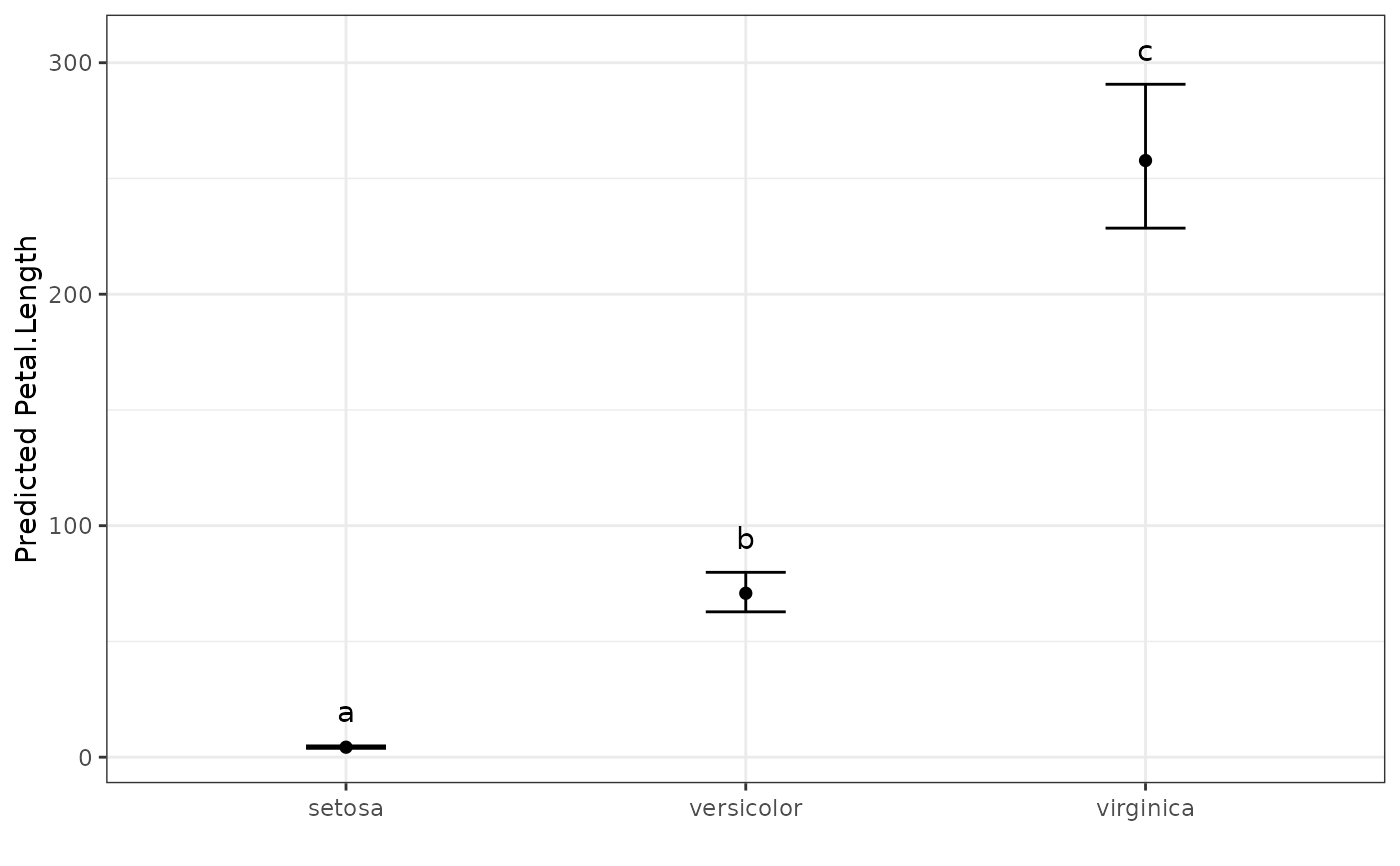
# Back transform, because the "original" data was exponential
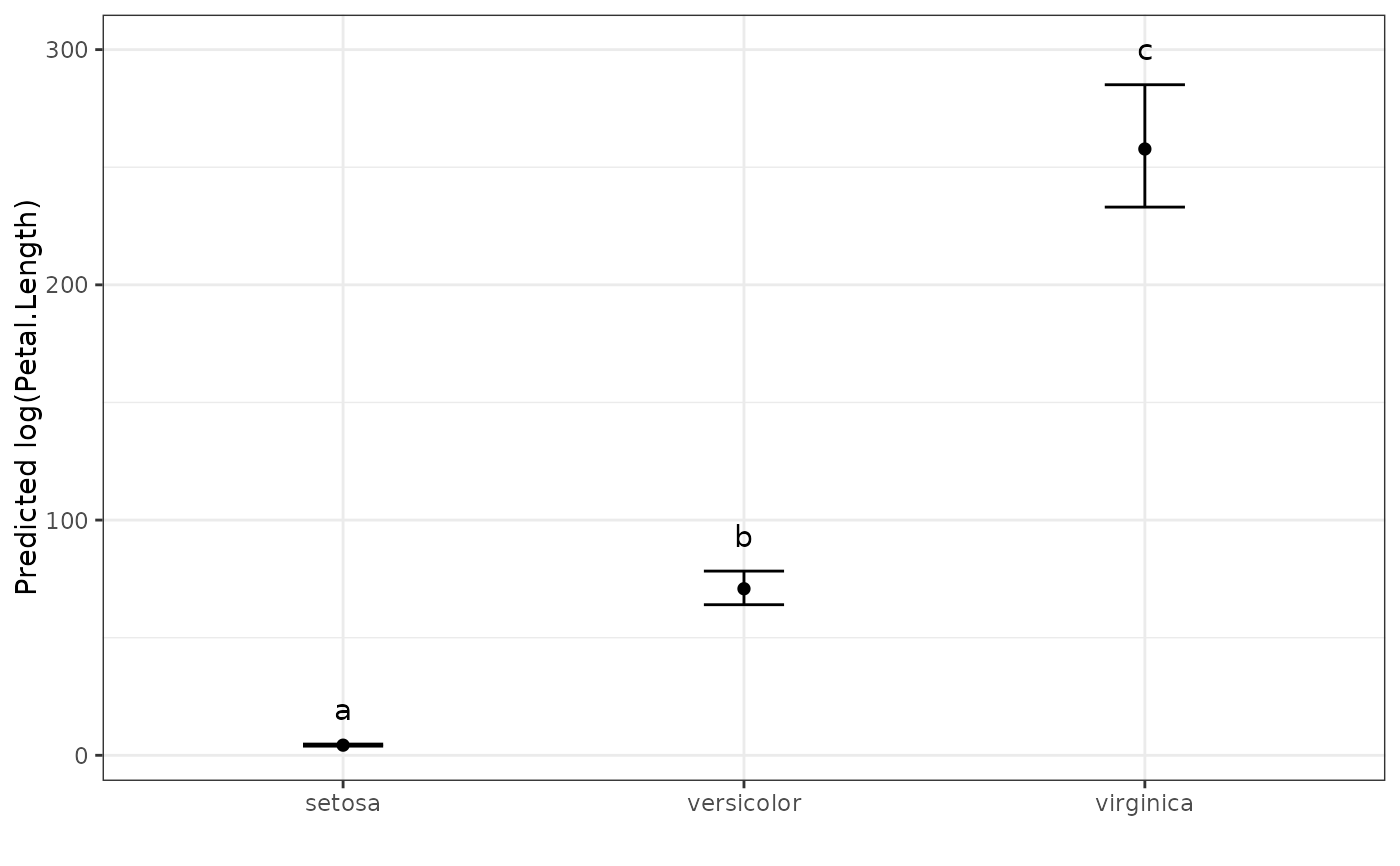
pred.out <- multiple_comparisons(log_model, classify = "Species",
trans = "log")
#> Warning: Offset value assumed to be 0. Change with `offset` argument.
# Display the predicted values table
pred.out
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: 0.20378
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci PredictedValue ApproxSE
#> 1 setosa 1.46 0.06 147 a 0.12 4.31 0.26
#> 2 versicolor 4.26 0.06 147 b 0.12 70.81 4.31
#> 3 virginica 5.55 0.06 147 c 0.12 257.75 15.69
#> low up
#> 1 3.83 4.87
#> 2 62.79 79.86
#> 3 228.54 290.69
# Show the predicted values plot
autoplot(pred.out, label_height = 15)
# Display the ANOVA table for the model
anova(log_model)
#> Analysis of Variance Table
#>
#> Response: log(Petal.Length)
#> Df Sum Sq Mean Sq F value Pr(>F)
#> Species 2 437.10 218.551 1180.2 < 2.2e-16 ***
#> Residuals 147 27.22 0.185
#> ---
#> Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
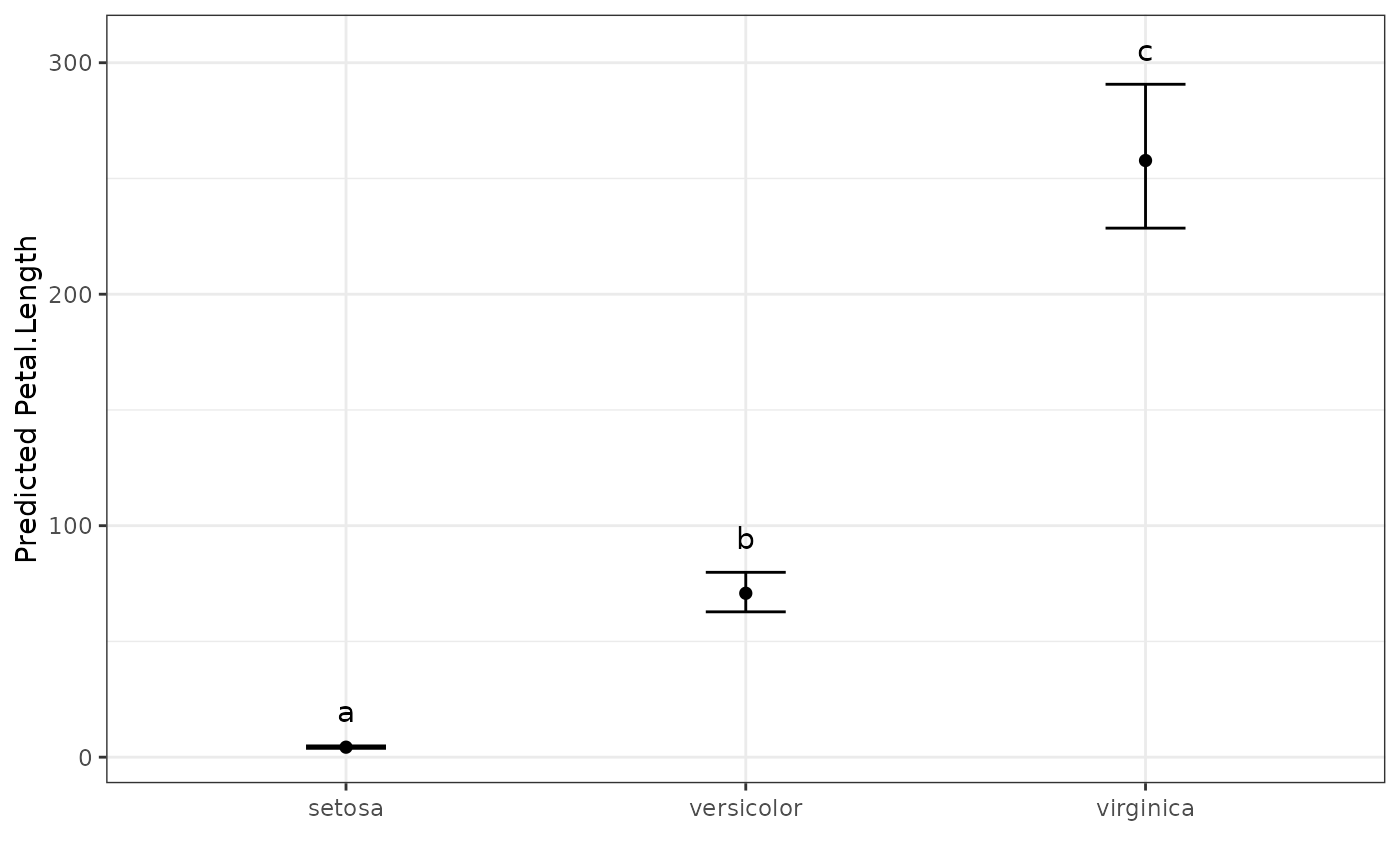
# Back transform, because the "original" data was exponential
pred.out <- multiple_comparisons(log_model, classify = "Species",
trans = "log")
#> Warning: Offset value assumed to be 0. Change with `offset` argument.
# Display the predicted values table
pred.out
#> Multiple Comparisons of Means: Tukey's HSD Test
#> Significance level: 0.05
#> HSD value: 0.20378
#>
#> Predicted values:
#> Species predicted.value std.error df groups ci PredictedValue ApproxSE
#> 1 setosa 1.46 0.06 147 a 0.12 4.31 0.26
#> 2 versicolor 4.26 0.06 147 b 0.12 70.81 4.31
#> 3 virginica 5.55 0.06 147 c 0.12 257.75 15.69
#> low up
#> 1 3.83 4.87
#> 2 62.79 79.86
#> 3 228.54 290.69
# Show the predicted values plot
autoplot(pred.out, label_height = 15)
 if (FALSE) { # \dontrun{
# ASReml-R Example
library(asreml)
# Fit ASReml-R model
model.asr <- asreml(yield ~ Nitrogen + Variety + Nitrogen:Variety,
random = ~ Blocks + Blocks:Wplots,
residual = ~ units,
data = asreml::oats)
wald(model.asr) # Nitrogen main effect significant
#Determine ranking and groups according to Tukey's Test
pred.out <- multiple_comparisons(model.obj = model.asr, classify = "Nitrogen",
descending = TRUE)
print(pred.out, decimals = 5)
# Example using a box-cox transformation
set.seed(42) # See the seed for reproducibility
resp <- rnorm(n = 50, 5, 1)^3
trt <- as.factor(sample(rep(LETTERS[1:10], 5), 50))
block <- as.factor(rep(1:5, each = 10))
ex_data <- data.frame(resp, trt, block)
# Change one treatment random values to get significant difference
ex_data$resp[ex_data$trt=="A"] <- rnorm(n = 5, 7, 1)^3
model.asr <- asreml(resp ~ trt,
random = ~ block,
residual = ~ units,
data = ex_data)
resplot(model.asr)
# lambda = 1/3 from MASS::boxcox()
model.asr <- asreml(resp^(1/3) ~ trt,
random = ~ block,
residual = ~ units,
data = ex_data)
resplot(model.asr) # Look much better
pred.out <- multiple_comparisons(model.obj = model.asr,
classify = "trt",
trans = "power", power = (1/3))
pred.out
autoplot(pred.out, label_height = 0.5)
} # }
if (FALSE) { # \dontrun{
# ASReml-R Example
library(asreml)
# Fit ASReml-R model
model.asr <- asreml(yield ~ Nitrogen + Variety + Nitrogen:Variety,
random = ~ Blocks + Blocks:Wplots,
residual = ~ units,
data = asreml::oats)
wald(model.asr) # Nitrogen main effect significant
#Determine ranking and groups according to Tukey's Test
pred.out <- multiple_comparisons(model.obj = model.asr, classify = "Nitrogen",
descending = TRUE)
print(pred.out, decimals = 5)
# Example using a box-cox transformation
set.seed(42) # See the seed for reproducibility
resp <- rnorm(n = 50, 5, 1)^3
trt <- as.factor(sample(rep(LETTERS[1:10], 5), 50))
block <- as.factor(rep(1:5, each = 10))
ex_data <- data.frame(resp, trt, block)
# Change one treatment random values to get significant difference
ex_data$resp[ex_data$trt=="A"] <- rnorm(n = 5, 7, 1)^3
model.asr <- asreml(resp ~ trt,
random = ~ block,
residual = ~ units,
data = ex_data)
resplot(model.asr)
# lambda = 1/3 from MASS::boxcox()
model.asr <- asreml(resp^(1/3) ~ trt,
random = ~ block,
residual = ~ units,
data = ex_data)
resplot(model.asr) # Look much better
pred.out <- multiple_comparisons(model.obj = model.asr,
classify = "trt",
trans = "power", power = (1/3))
pred.out
autoplot(pred.out, label_height = 0.5)
} # }